先做个广告:如需代注册帐号或代充值Chatgpt Plus会员,请添加站长客服微信:pingzi7749
论文:Empowering Beginners in Bioinformatics with ChatGPT. 2023
众所周知,与ChatGPT互动,给予的指令越精确,那么它给出的答案越精准。这篇论文提出一个与ChatGPT互动的模型:OPTICAL。其基本思想是通过迭代不断优化给予ChatGPT的指令。
该模型的流程图如下:
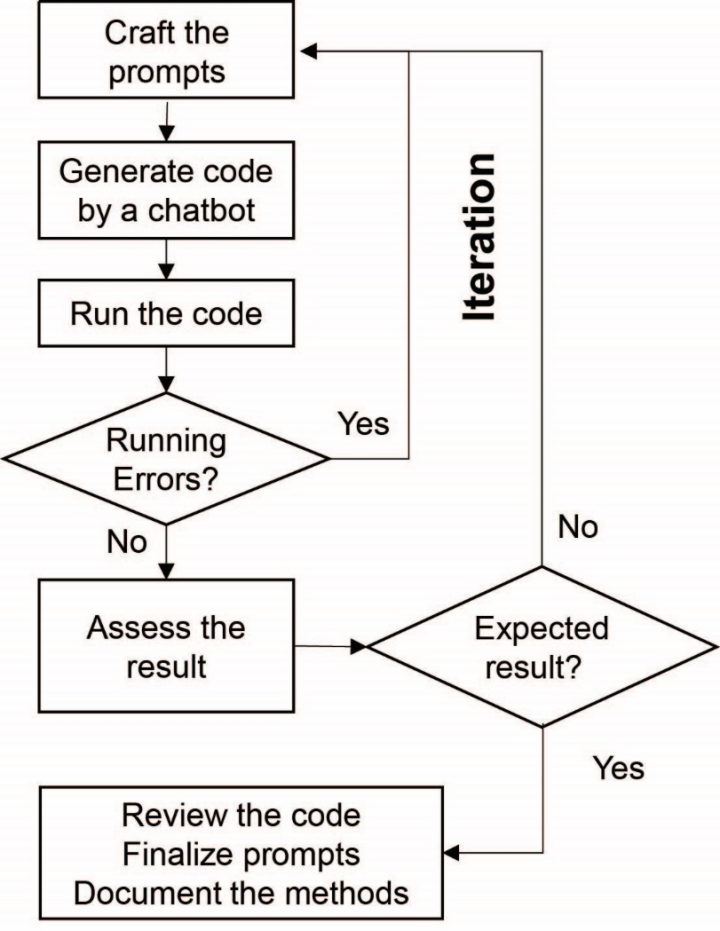
给予初始提示。
机器人产生分析代码。
运行代码。
如果出现错误,转向优化提示词。
如果代码正确,继续下一步。
评估结果。
如果结果不符合预期,转向优化提示词。
如果结果符合预期,继续下一步。
审查代码,得到最终提示词并归档方法。
迭代0
I have two fastq files in current folder from single-end sequencing of a ChIP-Seq library: ENCFF000AVS_1m.fastq.gz, and ENCFF000AVS_10m.fastq.gz. For each fastq file, align reads to the human reference genome, save to bam file, and then covert it to bigwig file. Tools to use: bowtie2, samtools, and deepTools. The index for bowtie2 is in the folder “../data/indx/bowtie2_whole_genome/” with “hg38” as the prefix. Use 24 CPU for the alignment. Please draft the code in bash.
迭代1
[E::idx_find_and_load] Could not retrieve index file for 'ENCFF000AVS_1m.bam'
迭代2
Wait, I saw that you have "samtools index" before "bamcoverage". Does bamcoverage as bam to be sorted before using as input?
审查代码
I need to insert line-by-line comments to the below code which works well to address the needs for the data analysis task. Wait for my code.
最终提示词(粗体字是经过迭代加入的提示细节):
Act as an experienced bioinformatician proficient in ChIP-Seq data analysis, you will assist me by writing code with number of lines as minimal as possible. Rest the thread if asked to. Reply “YES” if understand.
I have two fastq files in current folder from single-end sequencing of a ChIP-Seq library: ENCFF000AVS_1m.fastq.gz, and ENCFF000AVS_10m.fastq.gz. For each fastq file, align reads to the human reference genome, save to bam file, index it, and then covert it to bigwig file with CPM normalization. Tools to use: bowtie2, samtools, and deepTools. The index for bowtie2 is in the folder “../data/indx/bowtie2_whole_genome/” with “hg38” as the prefix. Use 24 CPU for the alignment. Please draft the code in bash.
安全二:推断DNA序列的分子进化系统发育树
定义聊天机器人的行为:
迭代0
迭代1
迭代2
迭代3
I wrote an R program to read a multiple alignment file named as tp53.clustal in ClustalW format, calculate evolutionary distance, build a NJ tree, and visualize the phylogeny. But I want to root the tree with the Zebrafish sequence as the outgroup. Can you help me revise the R code? Below is my R code.
# Load the required packageslibrary(seqinr)library(ape)# Read in the alignment filealn "tp53.clustal", format="clustal")# Calculate the evolutionary distancedist as.DNAbin(aln))# Build the NJ treetree # Plot the phylogenyplot(tree)
迭代4
迭代5
I got an error message complaining "Error in if (newroot == ROOT) { : argument is of length zero". Please fix it.
审查代码
I created the following R code. Please add inline comments.
最终提示词
无。
一键分析10X单细胞数据(点击图片跳转)
一键分析Bulk转录组数据(点击图片跳转)
本文链接:https://www.vipbxr.vip/GPT5_369.html
GPTs机器人GPTs机器人GPT-4.5 TurboGPT4.5GPT4.5官网GPT4.5发布GPT4.5网址GPT4.5网页版GPT4.5入口





网友评论